TogoImputation (beta) チュートリアル4、HIBAG を用いた HLA Genotype Imputation
Guacamole 環境への接続
この作業は、遺伝研個人ゲノム解析区画 Guacamole 環境から行いますので、VPN 接続を行い Guacamole に接続してください。
次に Guacamole デスクトップ環境の「アクティビティ」からターミナルを起動してください。
以下は上記で開いた Guacamole のデスクトップ中の Firefox や、そのデスクトップ内のターミナルの中で作業を行います。
システム利用方法
本システムでは、以下の流れで HIBAG を用いた HLA Genotype Imputation ワークフローを実行します。
- テスト入力データの準備
- HLA Genotype Imputation Workflow 用の設定ファイルの生成
- HLA Genotype Imputation Workflow の実行
テスト入力データの準備
チュートリアルをすす��めるにあたって、使用するテスト入力データをダウンロードし、遺伝研スパコン個人ゲノム解析区画へコピーします。 本ワークフローへの入力として必要となるデータは、PLINK の bed、bim、fam ファイルです。
PLINK bed、bim、fam ファイル の準備
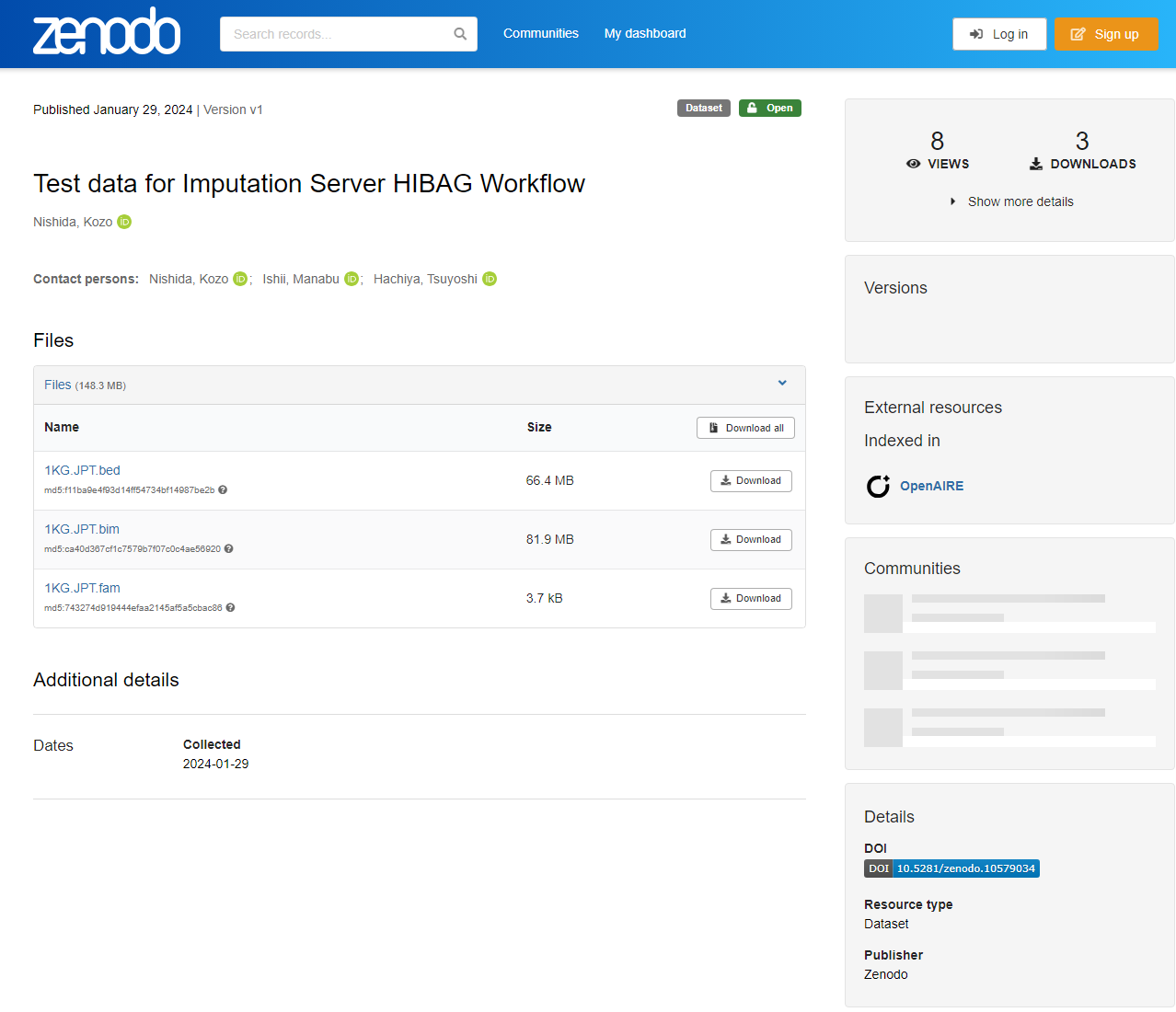
Test data for Imputation Server HIBAG Workflow にアクセスします。以下の3つのファイルがおいてあります。
1KG.JPT.bim1KG.JPT.fam1KG.JPT.bed
1KG.JPT.bim、1KG.JPT.fam、1KG.JPT.bed のすべてをダウンロードします。
遺伝研スパコン個人ゲノム解析区画へコピーします。
さきほどダウンロードしたテストデータをコピーします。
次に、さきほどダウンロードしたテストデータを次のコマンドでコピーします。(PLINKのファイルはすべて同じ directory 内に配置してください。)
以下の例では、コピーしたいテストデータは、ダウンロードフォルダの中にあり、コピー先��は、遺伝研スパコン個人ゲノム解析区画のお使いのアカウントのホームディレクトリになります。
scp -i 秘密鍵ファイル ~/Downloads/1KG.JPT.bim (お使いのアカウント名)@gwa.ddbj.nig.ac.jp:~/
scp -i 秘密鍵ファイル ~/Downloads/1KG.JPT.fam (お使いのアカウント名)@gwa.ddbj.nig.ac.jp:~/
scp -i 秘密鍵ファイル ~/Downloads/1KG.JPT.bed (お使いのアカウント名)@gwa.ddbj.nig.ac.jp:~/
これでテスト入力データの準備は終了です。
HLA Genotype Imputation Workflow 用の設定ファイルの生成
以下のアドレスにアクセスします。
http://localhost:5000/hibag
実際にアクセスすると、次のような画面になります。
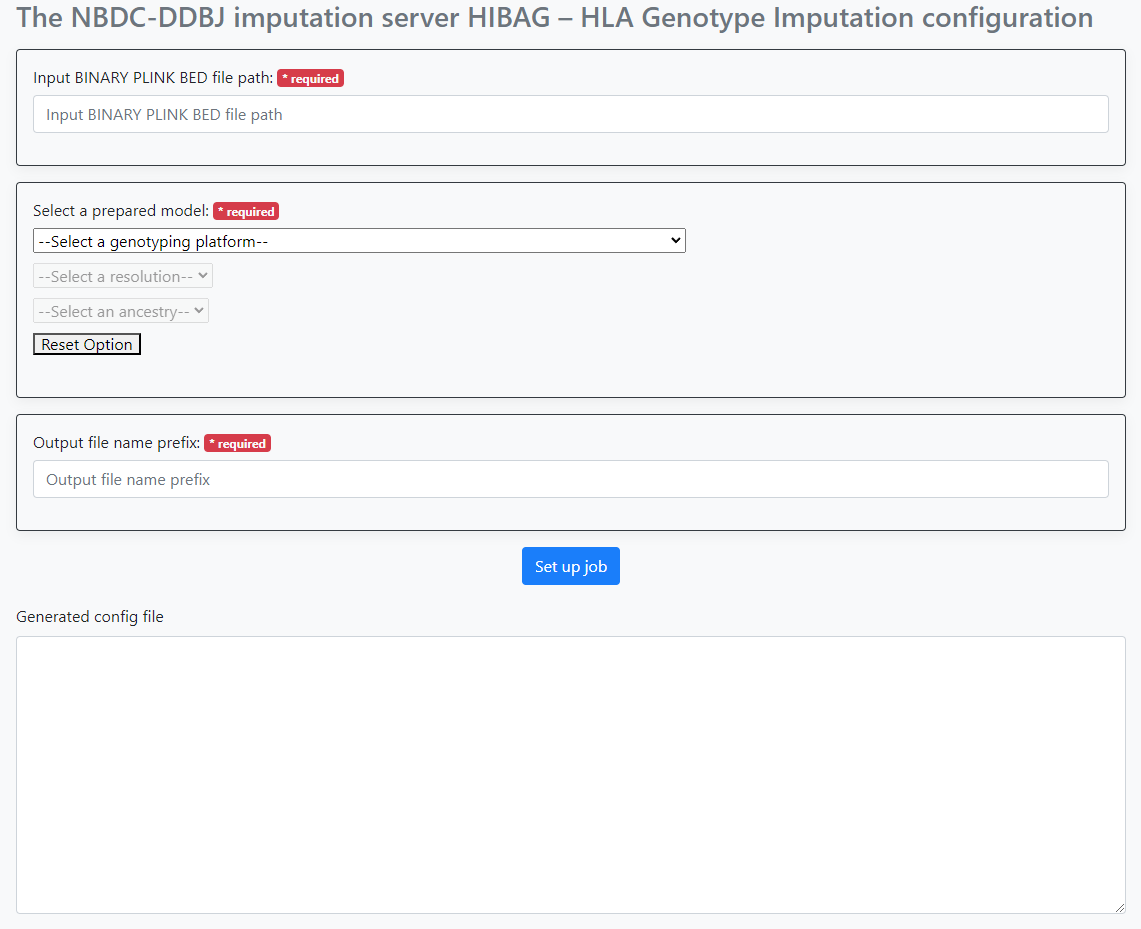
以下の項目について設定を行います。
- PLINK の bed ファイルのパス
- 本システムが予め備えている HIBAG のモデルの選択
- 本ワークフローが出力するファイルのプレフィックス名
「PLINK の bed ファイルのパス」 には、解析対象の bed ファイルのフルパスを指定します。
ここでは先程アップロードした、ファイルを使います。
具体的なフルパスは /home/youraccountname/1KG.JPT.bed のようになります。
次に HIBAG のモデルのオプションを選択します。 以下の3つについて、選択が可能です。3つの選択肢を選択すると、どのモデルを用いるかが決まります。
- Genotyping platform
- Resolution
- Ancestry
最後に 本ワークフローが出力するファイル(複数)のプレフィックス名 を入力します。
入力例は下記の画像ようになります。
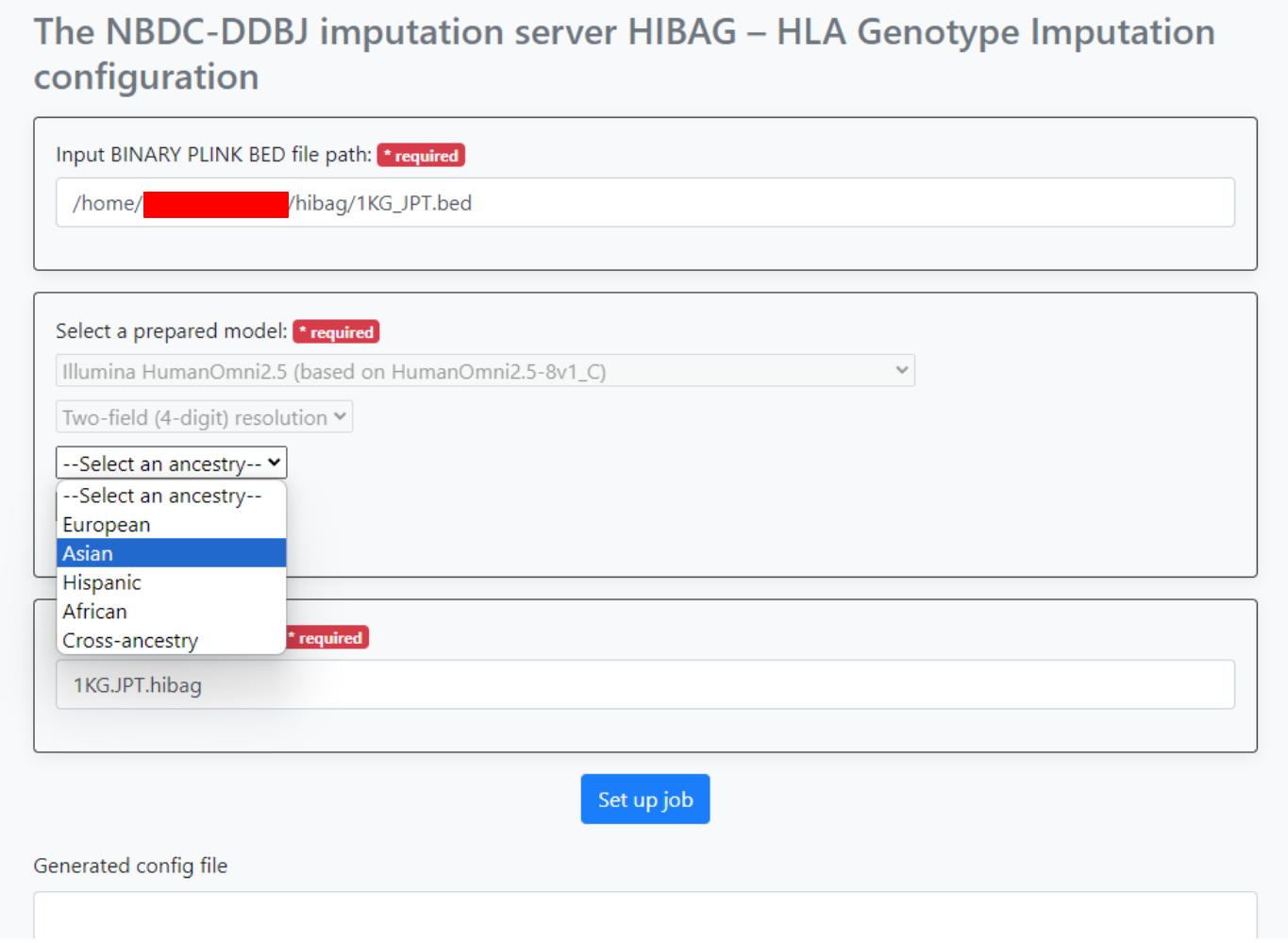
ここでは、入力の bed ファイルのパスを入力後、
--Select a genotyping platform--のドロップダウンリストでIllumina HumanOmni2.5 (based on HumanOmini2.5-8v1_Cを選択--Select a resolution--のドロップダウンリストでTwo-field (4-digit) resolutionを選択-Select an ancestry--のドロップダウンリストでAsianを選択
そして Output file name prefix に 1KG.JPT.hibag を入力しました。
(注意: Output file name prefix にパスを書くことはできません。 / が含まれているとエラーになります)
パラメータの指定が終わったら、Set up job ボタンを押します。 画面下部に、生成されたパラメータが表示されます。これをsapporo-web で使います。
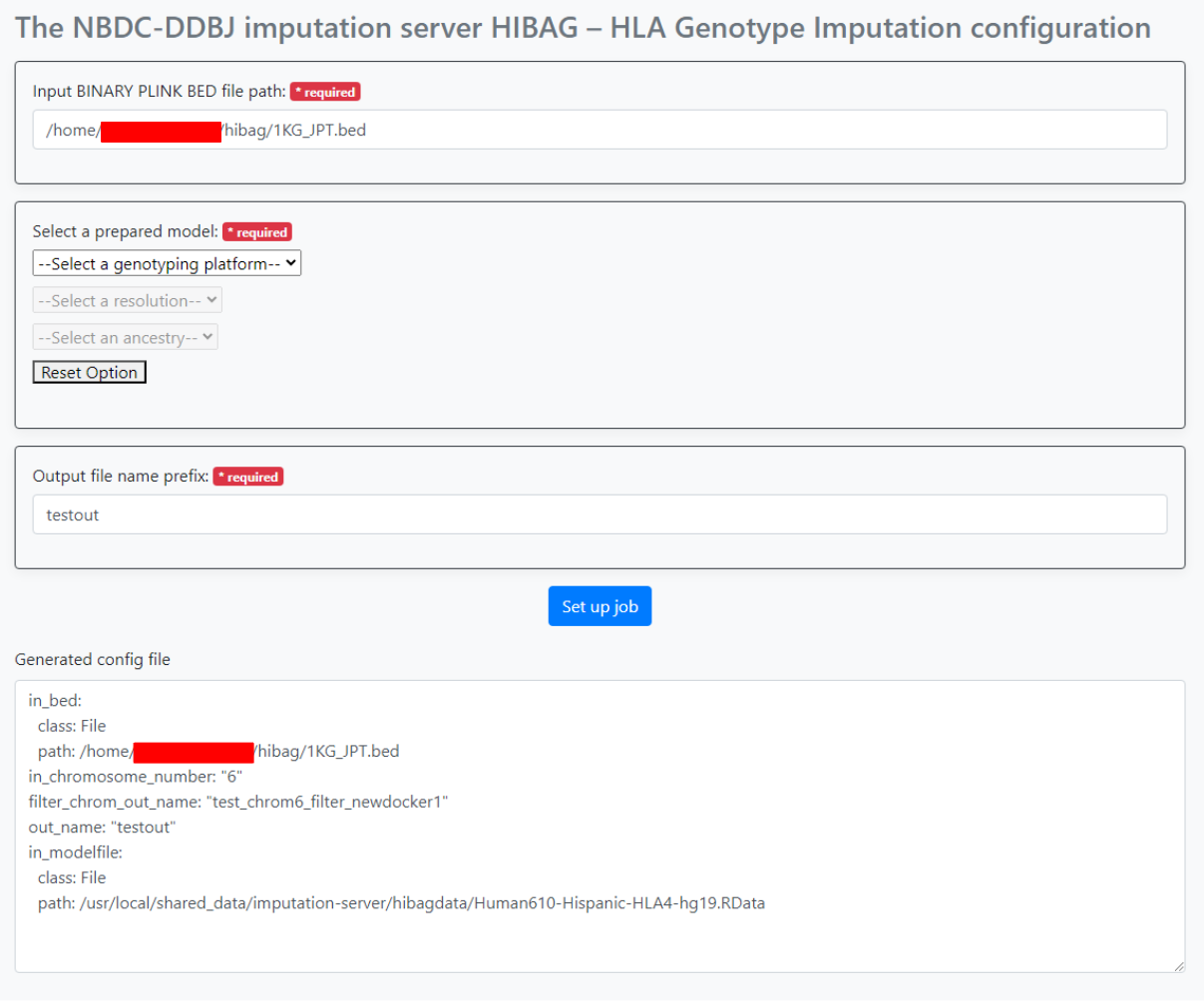
Imputation Workflowの実行
以下のアドレスにアクセスします。
http://localhost:1121
以下のような画面が表示されます。
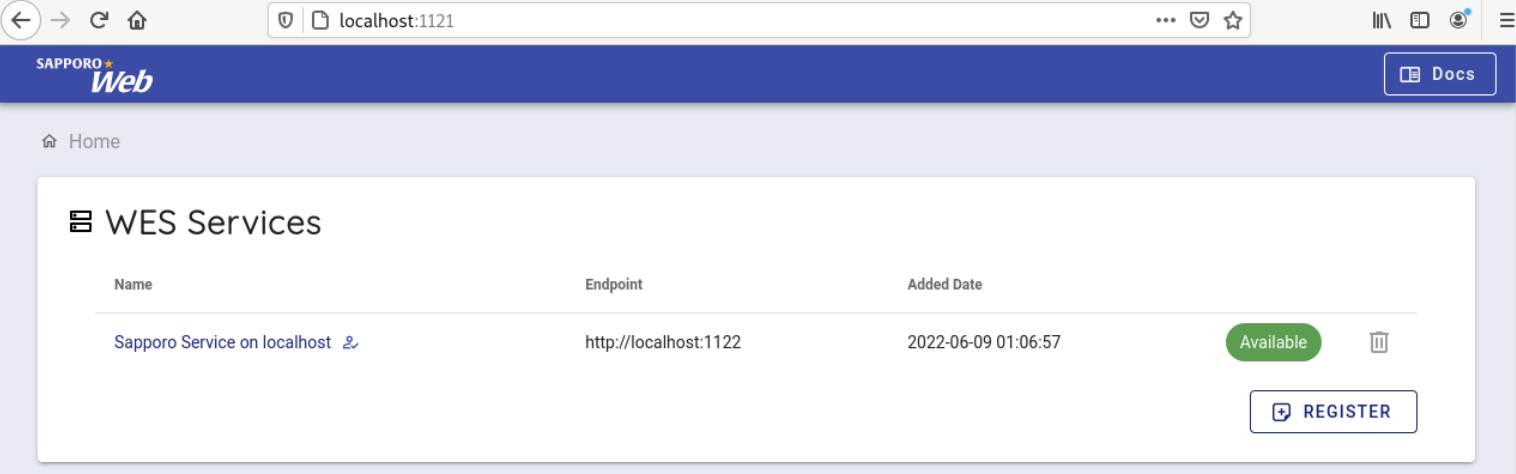
次に、デフォルトで使用可能になっている Sapporo Service on localhost を選択します。
クリックすると以下のような画面がでてきます
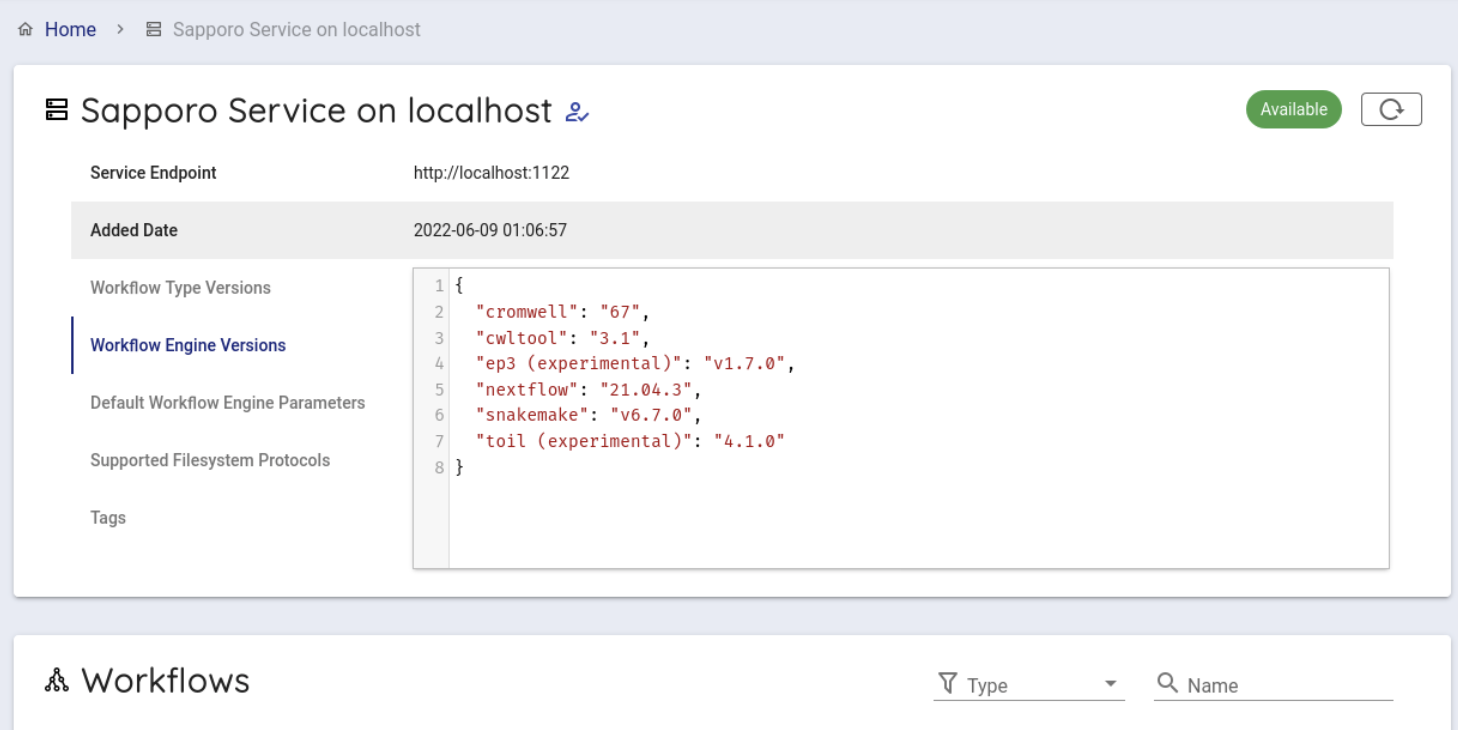
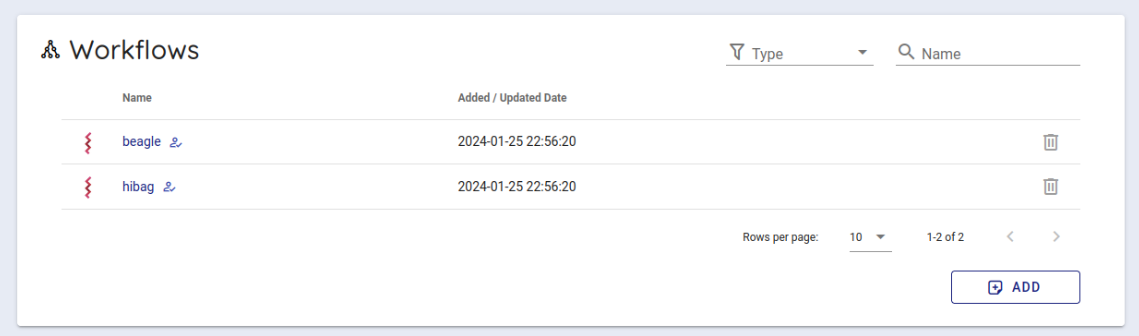
次にバックエンドワークフローを使用するために少し下にスクロールし、 Workflows という項目から hibag をクリックします。
Compose Run の項目から、Workflow Engine の項目で cwltool 3.1 を選択します。
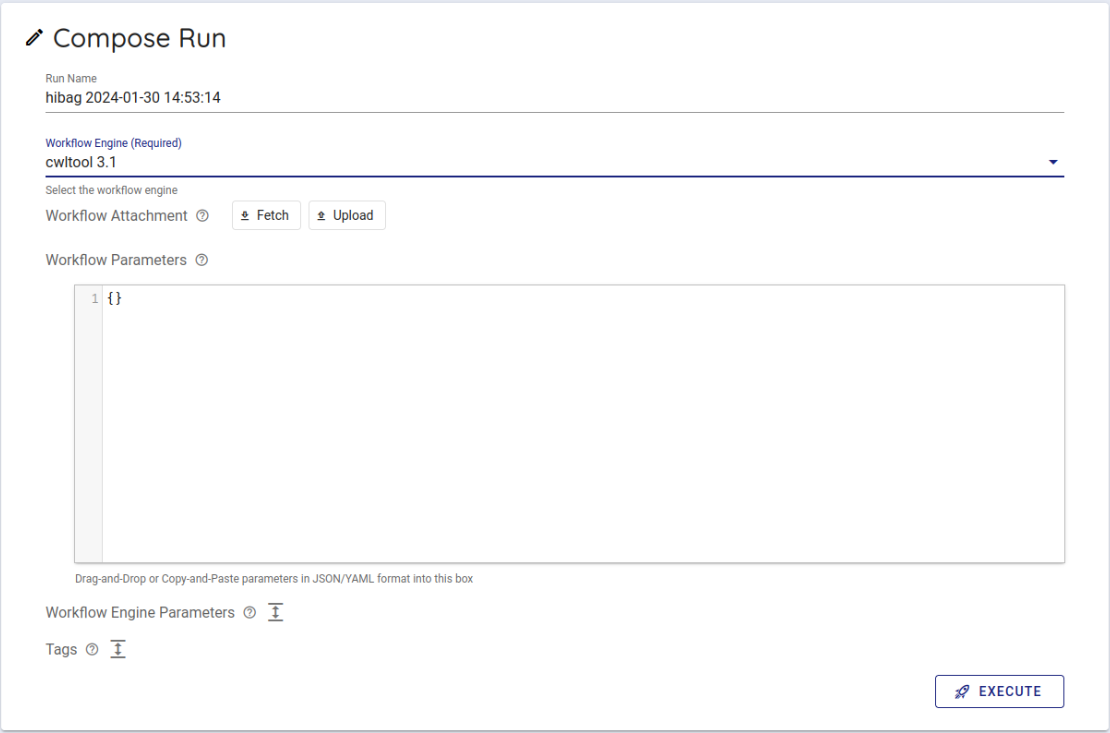
Workflow Parameters に先程、 imputationserver-web-ui で生成したパラメータを入力します。 このとき、最初から書かれている {} を消して、生成したパラメータを入力します。
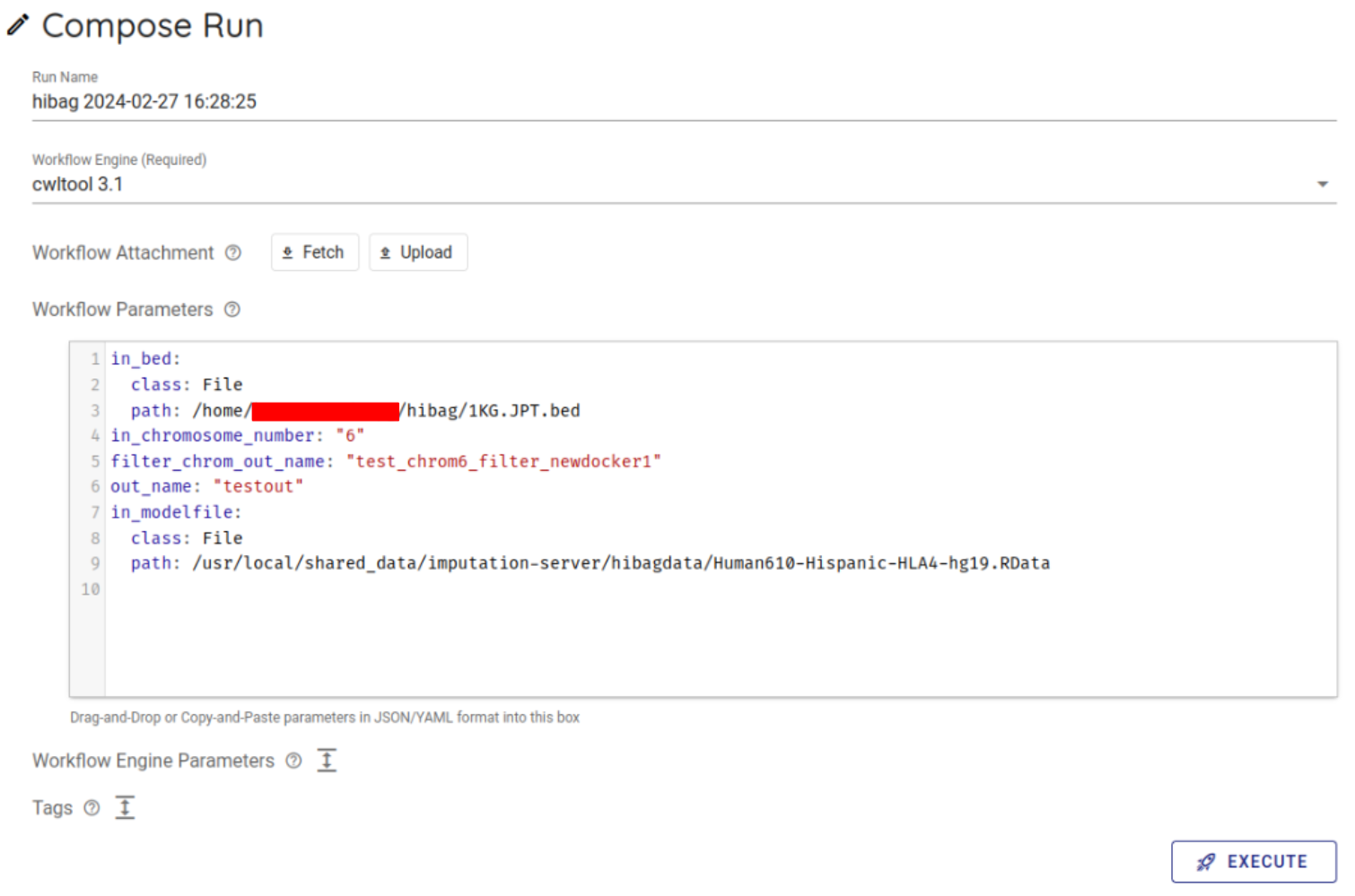
一番下にある EXECUTE ボタンを押して、ワークフローを実行します。 ジョブの状態が RUNNING になります。
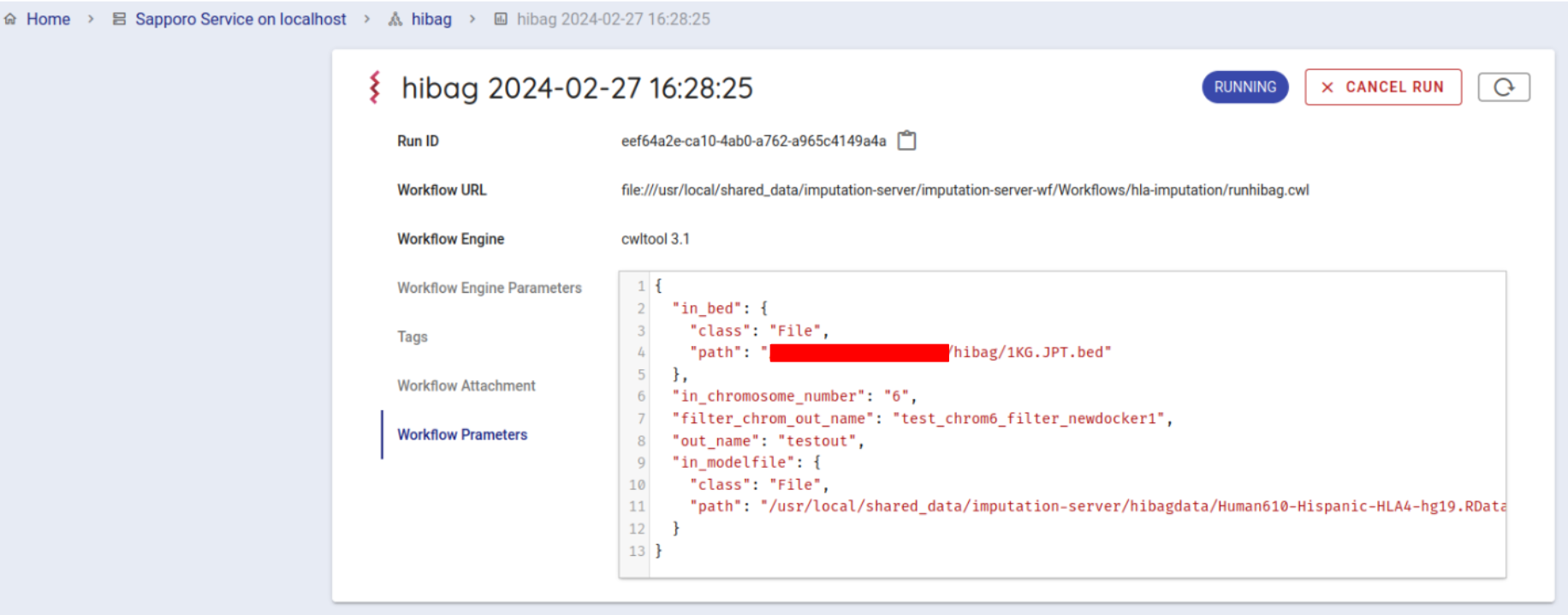
正常にワークフローの実行が開始されるとcwltoolでワークフローが実行されます。
正常に動作すると10分程度でステータスが COMPLETE に変わります。
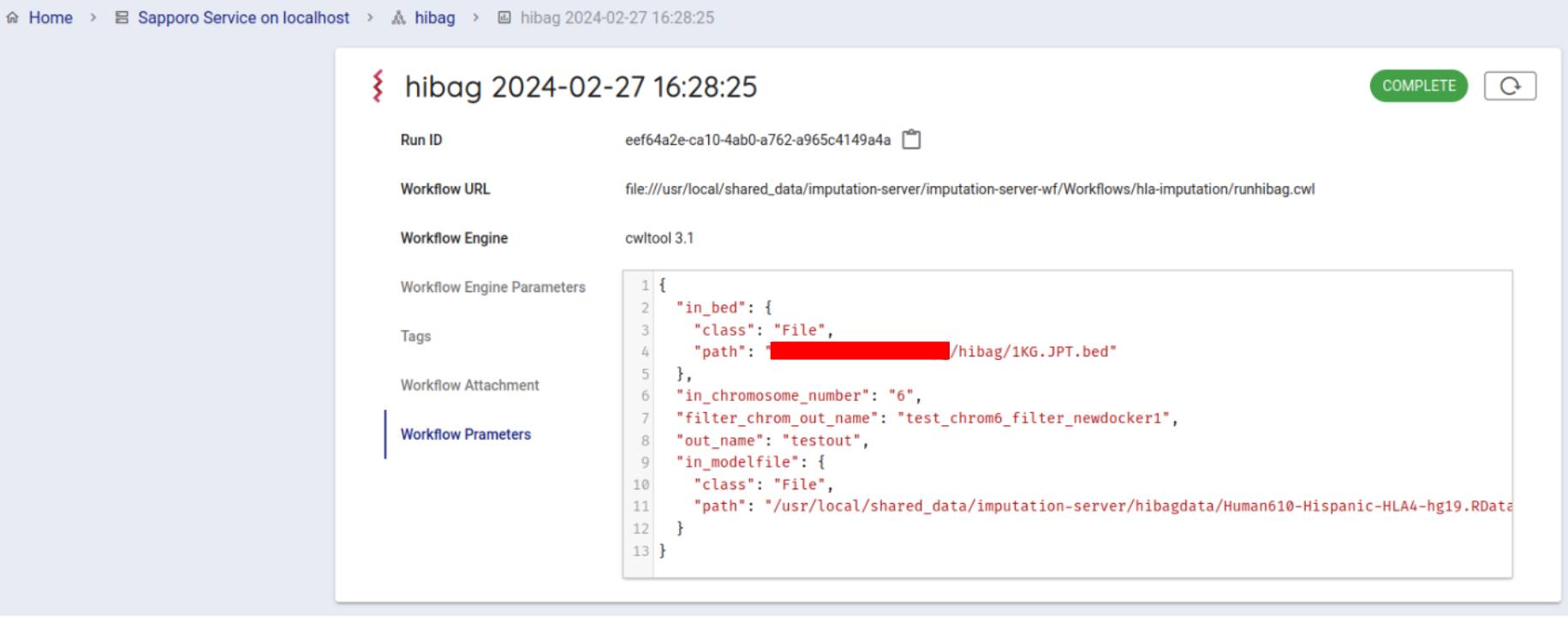
結果ファイルは、ブラウザから取得が可能です。 Run log の中の、Outputs をクリックすると結果ファイル一覧が表示されます。
ダウンロードしたいファイルをクリックするとダイアログが表示され、 デフォルトでは、 ~/ダウンロード 以下にダウンロードされます。
結果
Imputation Workflow 実行後、以下のものが取得できます。
ウェブブラウザから取得ができます。
以下のコマンドを、手元のパソコンにコピーすることが可能です。
ターミナルを開きます。
実行すると、現在コマンドを実行しているディレクトリにファイルがダウンロードされます。
scp (お使いのアカウント名)@gwa.ddbj.nig.ac.jp:~/ダウンロード/(ダウンロードしたいファイル名) .
(お使いのアカウント名)は、個人ゲノム解析環境へのログインに使用するアカウントです(ダウンロードしたいファイル名)に、ダウンロードしたいファイル名を指定します。
また、sapporo-serviceの結果ディレクトリから直接ダウンロードすることも可能です。
Run IDを調べます。Run ID の右に表示されているものが Run ID です。 右にあるアイコンをクリックすることで、 Run ID (以下runid)をコピーすることが可能です。

インストールしたディレクトリ/sapporo-service/run/runid の最初の2文字 /runid/outputs/ 以下にすべてのファイルがあります。
runidがeef64a2e-ca10-4ab0-a762-a965c4149a4aの場合、最初の2文字は ee になります。
scpでコピーするときは、お手元の計算機に以下のように入力します。 手元の計算機に、outputs というディレクトリが作成され、その中に解析結果が個人ゲノム解析区画から、お手元の計算機にコピーされてきます。
scp -i 秘密鍵ファイル -r (お使いのアカウント名)@gwa.ddbj.nig.ac.jp:~/sapporo-install/sapporo-service/run/ee/eef64a2e-ca10-4ab0-a762-a965c4149a4a/outputs outputs
作業を終える場合は、Guacamole を表示しているブラウザタブを閉じてください。