How to use NVIDIA Clara Parabricks
System Overview
NVIDIA Clara Parabricks is a genome analysis pipeline which is compatible with GATK. It is possible to significantly reduce the processing time compared to using the official implementation of GATK by using GPU.
References
Parabricks are installed and provided on GPU nodes managed under the Slurm resource scheduler in the personal genome analysis division of the NIG supercomputer, and jobs can be submitted using the advance reservations of the Slurm resource scheduler.
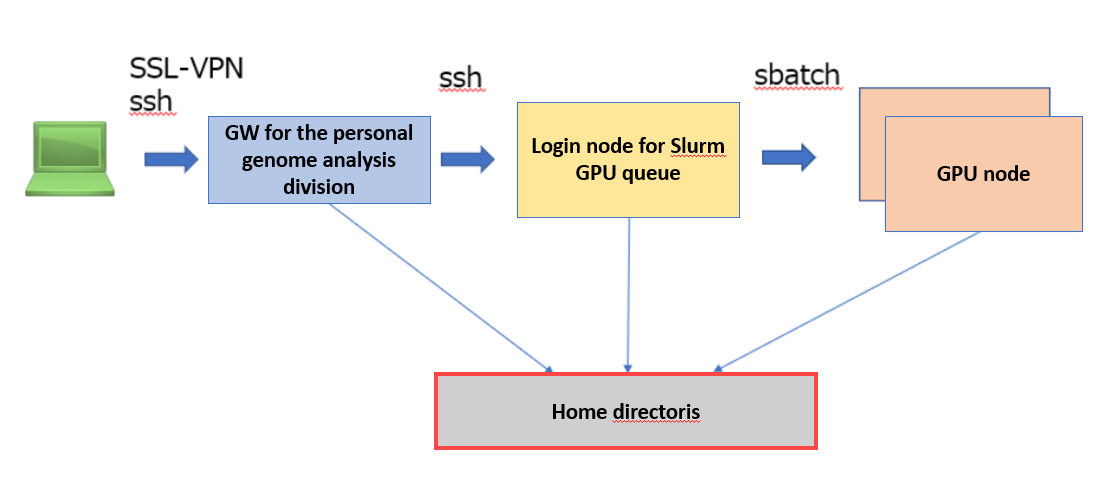
- The interactive node for the Slurm GPU queue is shared with other users. If you do not want to share the interactive node, submit with the normal procedure to rent a compute node in the personal genome analysis division and contact us to install Slurm on it.
- When using Parabricks, it is assumed that all GPUs (4 GPUs) of the GPU node are used, so that no other user's job can be on the same GPU node at the same time.
Use Procedure Overview
Here is the procedure for using Parabricks in the Personal Genome Analysis division of the NIG supercompute.
- Preparation for use
- apply for use of the Personal Genome Analysis division. Refer here for how to apply for use.
- submit the usage plan table.
- specify your advance reservation setting.
- Log in and submit jobs: log in to the Slurm interactive node and submit jobs to the worker node (GPU node).
How to fill in the usage plan table when using Parabricks
Prepare and submit a usage plan table as described on the page "Submission of Your Usage Plan".
The following is how to fill in the unadjusted trial balance for the priority use of GPU nodes in the Personal Genome Analysis division.
- Service: select 'Priority use of computing nodes_Personal genome analysis division'.
- Type: select 'Thin (Intel/NVIDIA GPU) (1 unit = 1GPU 4CPU cores 48GB memory).
- Specify the usage in units of 4.

For the start and end dates, enter the dates set using the advance reservation command. You may not be able to make a reservation on the desired dates during busy times, so replace your usage plan table with the reserved start and end dates when the reservation has been made.
How to specify the advance reservation
The basic procedure is the same as the advance reservation for Grid Engine in the general analysis division. (See Instruction for using Advance Reservation Service).
- The instruction for the advance reservation command by using Slurm is currently being developed, so contact us to reserve slots.
How to login to a Slurm interactive node
Please submit an application to use the Personal Genome Analysis division and to be granted login access to the parabricks analysis environment (at022vm02). Afterward, please log in to the gateway to the Personal Genome Analysis division and then log in to the parabricks analysis environment using the following command.
ssh at022vm02
How to submit a job
Build an analysis environment
To execute a GATK-compatible genome analysis pipeline on Parabricks, we can utilize the following workflow. https://github.com/NCGM-genome/WGSpipeline
To install the workflow for executing a GATK-compatible genome analysis pipeline on Parabricks, run the following command to clone the workflow.
$ cd /path/to/working/directory/
$ git clone https://github.com/NCGM-genome/WGSpipeline.git
Tutorial
This tutorial demonstrates how to execute the germline-gpu.cwl workflow, which you have installed from the WGSpipeline repository.
Download dataset for the tutorial
The tutorial dataset can be downloaded by executing the following command. The download links are provided here.
$ cd /path/to/working/directory/
$ OUTDIR=wgs_fastq ; mkdir -p $OUTDIR ; for url in `cat WGSpipeline/download_links/wgs_fastq_NA12878_20k.download_links.txt` ; do echo $url ; file=`basename $url` ; if [ ! -f ${OUTDIR}/$file ] ; then wget $url -O ${OUTDIR}/$file ; fi ; done
The tutorial reference and resouce can be downloaded by executing the following command. The download links are provided here.
$ cd /path/to/working/directory/
$ OUTDIR=reference_hg38 ; mkdir -p $OUTDIR ; for url in `cat WGSpipeline/download_links/reference_hg38.download_links.txt` ; do echo $url ; file=`basename $url` ; if [ ! -f ${OUTDIR}/$file ] ; then wget $url -O ${OUTDIR}/$file ; fi ; done
Tutorial 1: Execute germline-gpu.cwl using a single pair of FASTQ files
In Tutorial 1, we will execute germline-gpu.cwl using a single pair of FASTQ files. Create a script named /path/to/working/directory/tutorial1.sh with the following content:
#!/bin/bash
#
#SBATCH --partition=igt009
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=48
#SBATCH --job-name=tutorial1
#SBATCH --output=tutorial1.log
#SBATCH --mem 384000
cd /path/to/working/directory
mkdir -p tutorial_01
cwltool --singularity \
--outdir tutorial_01 \
WGSpipeline/Workflows/germline-gpu.cwl \
--ref reference_hg38/Homo_sapiens_assembly38.fasta \
--fq1 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06HDADXX130110.1\\tPL:ILLUMINA\\tPU:H06HDADXX130110.1\\tLB:H06HDADXX130110.1\\tSM:NA12878" \
--num_gpus 4 \
--prefix NA12878.H06HDADXX130110.1 \
--autosome_interval WGSpipeline/interval_files/autosome.bed \
--PAR_interval WGSpipeline/interval_files/PAR.bed \
--chrX_interval WGSpipeline/interval_files/chrX.bed \
--chrY_interval WGSpipeline/interval_files/chrY.bed
Subsequently, the analysis will be conducted using the following command.
$ cd /path/to/working/directory
$ sbatch tutorial1.sh
When the --knownSites option is omitted, an empty BQSR recalibration table (.bqsr.recal.table) will be generated.
Output files will be saved to the following directory.
/path/to/working/directory/tutorial_01
|--NA12878.H06HDADXX130110.1.PAR.g.vcf.gz
|--NA12878.H06HDADXX130110.1.PAR.g.vcf.gz.tbi
|--NA12878.H06HDADXX130110.1.autosome.g.vcf.gz
|--NA12878.H06HDADXX130110.1.autosome.g.vcf.gz.tbi
|--NA12878.H06HDADXX130110.1.bqsr.recal.table
|--NA12878.H06HDADXX130110.1.chrX_female.g.vcf.gz
|--NA12878.H06HDADXX130110.1.chrX_female.g.vcf.gz.tbi
|--NA12878.H06HDADXX130110.1.chrX_male.g.vcf.gz
|--NA12878.H06HDADXX130110.1.chrX_male.g.vcf.gz.tbi
|--NA12878.H06HDADXX130110.1.chrY.g.vcf.gz
|--NA12878.H06HDADXX130110.1.chrY.g.vcf.gz.tbi
|--NA12878.H06HDADXX130110.1.cram
|--NA12878.H06HDADXX130110.1.cram.crai
Tutorial 2: Execute germline-gpu.cwl using multiple pairs of FASTQ files
In Tutorial 2, we will execute germline-gpu.cwl using multiple pairs of FASTQ files.
The --fq1, --fq2, and --rg options can be specified multiple times. The number of --fq1 options must match the number of --fq2 and --rg options. Please ensure that these options are listed in the order of --fq1, --fq2, and --rg.
Create a script named /path/to/working/directory/tutorial2.sh with the following content:
#!/bin/bash
#
#SBATCH --partition=igt009
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=48
#SBATCH --job-name=tutorial2
#SBATCH --output=tutorial2.log
#SBATCH --mem 384000
cd /path/to/working/directory
mkdir -p tutorial_02
cwltool --singularity \
--outdir tutorial_02 \
WGSpipeline/Workflows/germline-gpu.cwl \
--ref reference_hg38/Homo_sapiens_assembly38.fasta \
--fq1 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06HDADXX130110.1\\tPL:ILLUMINA\\tPU:H06HDADXX130110.1\\tLB:H06HDADXX130110.1\\tSM:NA12878" \
--fq1 wgs_fastq/H06HDADXX130110.2.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06HDADXX130110.2.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06HDADXX130110.2\\tPL:ILLUMINA\\tPU:H06HDADXX130110.2\\tLB:H06HDADXX130110.2\\tSM:NA12878" \
--fq1 wgs_fastq/H06JUADXX130110.1.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06JUADXX130110.1.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06JUADXX130110.1\\tPL:ILLUMINA\\tPU:H06JUADXX130110.1\\tLB:H06JUADXX130110.1\\tSM:NA12878" \
--num_gpus 4 \
--prefix NA12878 \
--autosome_interval WGSpipeline/interval_files/autosome.bed \
--PAR_interval WGSpipeline/interval_files/PAR.bed \
--chrX_interval WGSpipeline/interval_files/chrX.bed \
--chrY_interval WGSpipeline/interval_files/chrY.bed
Subsequently, the analysis will be conducted using the following command.
$ cd /path/to/working/directory
$ sbatch tutorial2.sh
Output files will be saved to the following directory.
/path/to/working/directory/tutorial_02
|--NA12878.PAR.g.vcf.gz
|--NA12878.PAR.g.vcf.gz.tbi
|--NA12878.autosome.g.vcf.gz
|--NA12878.autosome.g.vcf.gz.tbi
|--NA12878.bqsr.recal.table
|--NA12878.chrX_female.g.vcf.gz
|--NA12878.chrX_female.g.vcf.gz.tbi
|--NA12878.chrX_male.g.vcf.gz
|--NA12878.chrX_male.g.vcf.gz.tbi
|--NA12878.chrY.g.vcf.gz
|--NA12878.chrY.g.vcf.gz.tbi
|--NA12878.cram
|--NA12878.cram.crai
Tutorial 3: Execute germline-gpu.cwl using --knownSites option
In Tutorial 3, we will execute germline-gpu.cwl using --knownSites option.
Create a script named /path/to/working/directory/tutorial3.sh with the following content:
#!/bin/bash
#
#SBATCH --partition=igt009
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=48
#SBATCH --job-name=tutorial3
#SBATCH --output=tutorial3.log
#SBATCH --mem 384000
cd /path/to/working/directory
mkdir -p tutorial_03
cwltool --singularity \
--outdir tutorial_03 \
WGSpipeline/Workflows/germline-gpu.cwl \
--ref reference_hg38/Homo_sapiens_assembly38.fasta \
--knownSites reference_hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz \
--knownSites reference_hg38/Homo_sapiens_assembly38.known_indels.vcf.gz \
--fq1 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06HDADXX130110.1.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06HDADXX130110.1\\tPL:ILLUMINA\\tPU:H06HDADXX130110.1\\tLB:H06HDADXX130110.1\\tSM:NA12878" \
--fq1 wgs_fastq/H06HDADXX130110.2.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06HDADXX130110.2.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06HDADXX130110.2\\tPL:ILLUMINA\\tPU:H06HDADXX130110.2\\tLB:H06HDADXX130110.2\\tSM:NA12878" \
--fq1 wgs_fastq/H06JUADXX130110.1.ATCACGAT.20k_reads_1.fastq \
--fq2 wgs_fastq/H06JUADXX130110.1.ATCACGAT.20k_reads_2.fastq \
--rg "@RG\\tID:NA12878.H06JUADXX130110.1\\tPL:ILLUMINA\\tPU:H06JUADXX130110.1\\tLB:H06JUADXX130110.1\\tSM:NA12878" \
--num_gpus 4 \
--prefix NA12878 \
--autosome_interval WGSpipeline/interval_files/autosome.bed \
--PAR_interval WGSpipeline/interval_files/PAR.bed \
--chrX_interval WGSpipeline/interval_files/chrX.bed \
--chrY_interval WGSpipeline/interval_files/chrY.bed
Subsequently, the analysis will be conducted using the following command.
$ cd /path/to/working/directory
$ sbatch tutorial3.sh
With the --knownSites option, a non-empty BQSR table file (.bqsr.recal.table) will be generated. However, please note that the output cram file will not have undergone BQSR recalibration.
Output files will be saved to the following directory.
/path/to/working/directory/tutorial_03
|--NA12878.PAR.g.vcf.gz
|--NA12878.PAR.g.vcf.gz.tbi
|--NA12878.autosome.g.vcf.gz
|--NA12878.autosome.g.vcf.gz.tbi
|--NA12878.bqsr.recal.table
|--NA12878.chrX_female.g.vcf.gz
|--NA12878.chrX_female.g.vcf.gz.tbi
|--NA12878.chrX_male.g.vcf.gz
|--NA12878.chrX_male.g.vcf.gz.tbi
|--NA12878.chrY.g.vcf.gz
|--NA12878.chrY.g.vcf.gz.tbi
|--NA12878.cram
|--NA12878.cram.crai
Reference: usage for germline-gpu.cwl
usage: Workflows/germline-gpu.cwl [-h] [--bwa_options STRING] \
--ref FILE \
[--knownSites FILE] \
--fq1 FILE \
--fq2 FILE \
--rg STRING \
--autosome_interval FILE \
--PAR_interval FILE \
--chrX_interval FILE \
--chrY_interval FILE \
--num_gpus INT \
--prefix STRING
optional arguments:
-h, --help Show this help message and exit.
--bwa_options STRING Pass supported bwa mem options as one string.
The current original bwa mem supported options are -M, -Y, and -T.
(e.g. --bwa-options="-M -Y")
(default: "-T 0 -Y")
--ref FILE Path to the reference file.
--knownSites FILE Path to a known indels file.
The file must be in vcf.gz format.
This option can be used multiple times.
--fq1 FILE Path to FASTQ file 1.
This option can be used multiple times.
--fq2 FILE Path to FASTQ file 2.
This option can be used multiple times.
--rg STRING Read group string.
This option can be used multiple times.
--autosome_interval FILE Path to interval BED file for autosome regions.
--PAR_interval FILE Path to interval BED file for PAR regions.
--chrX_interval FILE Path to interval BED file for chrX regions.
--chrY_interval FILE Path to interval BED file for chrY regions.
--num_gpus INT Number of GPUs to use for a run (should be ≥1).
--prefix STRING Output file prefix.

