Parallel Jobs
When running programs that utilize multiple CPU cores simultaneously for extended periods, please execute them as parallel jobs. For executing many such programs, use array jobs for parallel tasks. The submission command should utilize the sbatch command.
Slurm does not have a feature equivalent to the Parallel Environment (PE) in AGE. We will introduce how to specify options to allocate cores in a nearly equivalent manner.
Reference Information:
- Support for MultiCore/Multi-Thread Architecture (detailed documentation from the developer)
- CPU Management User and Administrator Guide
Types of Parallel Jobs (Overview)
In line with the environments provided by PE in AGE, we present the options in Slurm to achieve nearly equivalent core allocations.
| Environment Name in AGE PE | Meaning of the PE Environment | Slurm Options to Specify for Similar Resource Allocation (Example) |
|---|---|---|
| def_slot | Secures NTASK number of CPU cores on the same compute node (The job will not start if NTASK exceeds the CPU cores available on the compute node) | -N 1-1 --n NTASK |
| mpi | Prepares NTASK number of CPU cores across multiple compute nodes, distributing tasks across nodes using round-robin | [-N NODES] -n NTASK --spread-job |
| mpi-fillup | Secures NTASK number of CPU cores across multiple compute nodes, ensuring as few nodes as possible are used | [-N NODES] -n NTASK |
| pe_n | Secures NTASK_1 number of CPU cores across multiple compute nodes, allocating NTASK_2 number of CPU cores per node | [-N NODES] -n NTASK_1 --ntasks-per-node=NTASK_2 |
Unlike the AGE PE, you cannot specify a range for NTASK. However, for NODES, a range can be specified, e.g., -N MINNODES-MAXNODES.
In Slurm, it's common to adjust the number of nodes with -N and the number of tasks per node with --ntasks-per-node to execute parallel jobs. Note that the product of the number of nodes and tasks per node equals the total number of tasks (parallelism).
Attention for Memory Request in Parallel Jobs
When specifying memory for parallel jobs with --mem (the memory allocated to the job per node) and --mem-per-cpu (the memory allocated to the job per CPU core), be aware that the system requests a memory capacity multiplied by the specified number of nodes or CPU cores.
Types of Parallel Jobs (Details)
CPU and Memory Allocation
When executing sbatch or srun commands without explicitly specifying the memory amount, the system allocates 8GB of memory per CPU core by default. This allocation can vary depending on the type of computer or queue used.
For example, specifying the following would allocate a total memory of 128GB on one compute node for the parallel job. Carefully consider this when deciding on the required memory amount.
-N 1-1 -n 16 --mem-per-cpu 8G
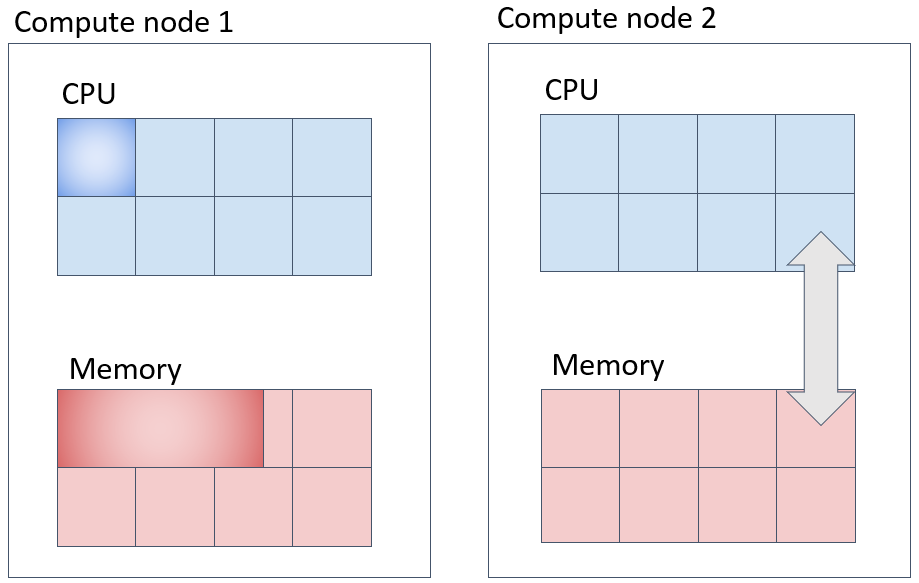
Parallel Job (1) (Corresponding to def_slot) - Method to Secure Specified Number of Cores on the Same Compute Node
To submit a job that acquires NTASK cores on a single compute node, use the following approach. Be mindful that specifying the --mem-per-cpu option will allocate memory based on the product of the CPU core count and the specified memory amount.
#!/bin/bash
#SBATCH -N 1-1
#SBATCH -n 2
#SBATCH --mem-per-cpu=20G
#SBATCH -t 0-00:10:00
#SABATCH -J an_example
make -j 2
In the example above, a total of 40GB of memory is allocated within one machine, with 20GB per task for 2 cores.
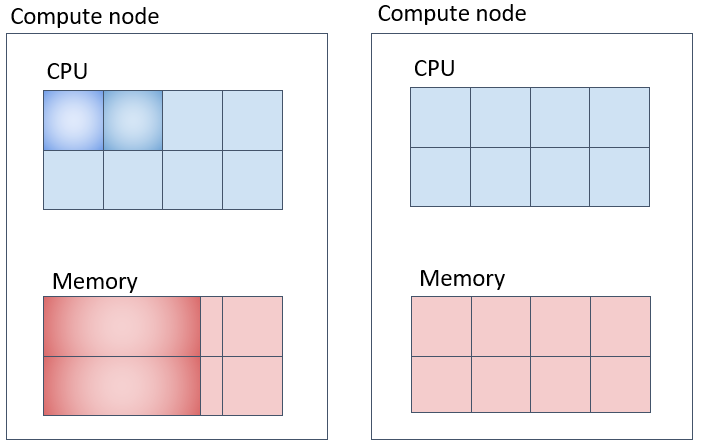
Parallel Job (2) (Corresponding to mpi) - Securing Specified Number of Cores on Different Compute Nodes as Much as Possible
#!/bin/bash
#SBATCH -N 2
#SBATCH -n 8
#SBATCH --spread-job
#SBATCH --mem-per-cpu=10G
#SBATCH -t 0-00:10:00
#SABATCH -J an_example
your_program
In this case, -N specifies the number of nodes to secure, -n is the total number of tasks for the job, and --spread-job tries to allocate an even number of tasks across the nodes.
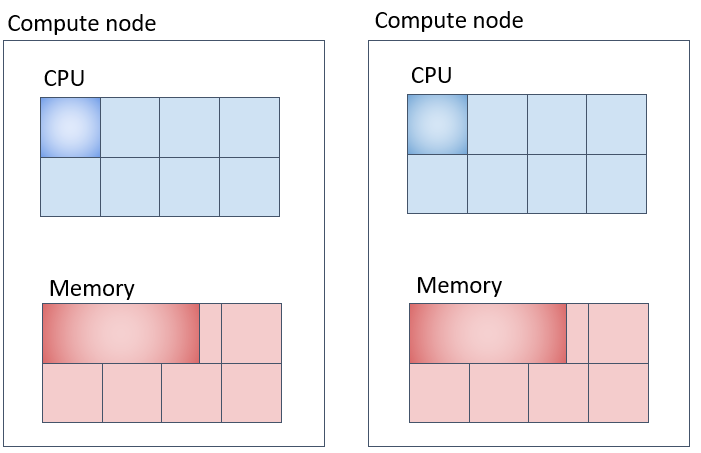
Parallel Job (3) (Corresponding to mpi-fillup) - Securing Specified Number of Cores on the Same Compute Node as Much as Possible
Slurm is configured to fill up compute nodes with tasks as much as possible.
#!/bin/bash
#SBATCH -N 2
#SBATCH -n 4
#SBATCH -t 0-00:10:00
#SBATCH --mem-per-cpu=20G
#SABATCH -J an_example
your_program
By default, Slurm allocates tasks while filling up compute nodes. If it cannot fit all tasks, it seeks resources on another machine. In the example above, one machine is allocated 3 cores × 20GB = 60GB, and the other 1 core × 20GB = 20GB, totaling 80GB for 4 cores.
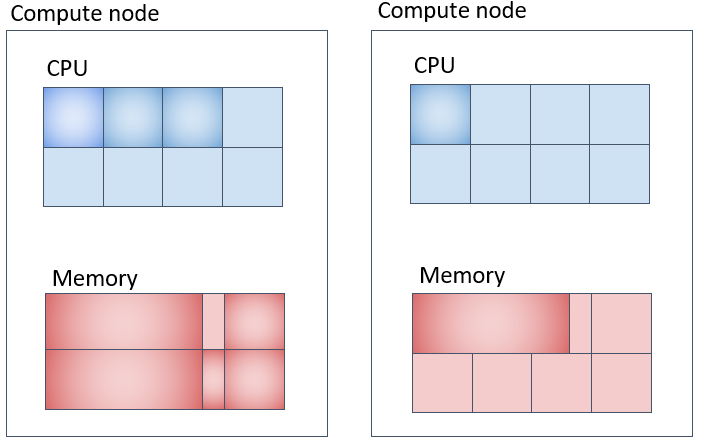
Parallel Job (4) (Corresponding to pe_n) - Using Multiple Compute Nodes by Securing Specified Number of Cores on Each Node
#!/bin/bash
#SBATCH -N 2
#SBATCH -n 8
#SBATCH -t 0-00:10:00
#SBATCH --ntasks-per-node=4
#SBATCH --mem-per-cpu=8G
#SABATCH -J an_example
your program
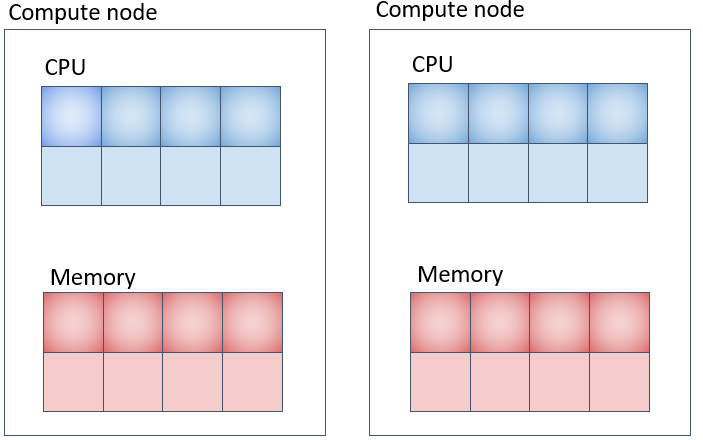
The memory allocation in the example above is 8GB per CPU core, totaling 32GB per machine, and 64GB across 2 machines.
Executing MPI Programs
The supercomputer at the Genetics Research Institute uses OpenMPI as the MPI implementation. Use the OpenMPI mpirun command to launch MPI program execution modules. With mpirun and Slurm combination, it's unnecessary to specify options like -np for mpirun.
Executing Programs Written in OpenMP
When executing binary modules of programs using OpenMP, you can specify the OMP_NUM_THREADS as an environment variable to set the number of threads for parallel execution within a node. It's possible to omit the OMP_NUM_THREADS specification, but by default, the program will attempt to use all recognized CPU cores. If running a job exclusively on one compute node, it's fine to not specify OMP_NUM_THREADS. However, if sharing a compute node among multiple jobs or when multiple options affecting the environment are specified and it's unclear which value will be set, explicitly setting OMP_NUM_THREADS is recommended.
#!/bin/bash
#SBATCH -N 1-1
#SBATCH -n 1
#SBATCH -c 10
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
./omp_sample
exit $?
When specified as above, the following meanings apply:
-N 1-1specifies the number of nodes to be used for the job.-n 1specifies the number of tasks to be launched for the job.-c 10specifies the number of CPU cores to be used for the job. This number is assigned to the SLURM_CPUS_PER_TASK environment variable.
Occupying a single compute node
#!/bin/bash
#SBATCH -N 1-1
#SBATCH -n 1
#SBATCH --exclusive
./omp_sample
exit $?
Slurm environment variables used for parallel jobs
The following environment variables are passed to the environment variables of the job when it is executed. These can be used to instruct the internal parallel behaviour as shown in the example above. For detailed information on available environment variables, refer to the online manual.
| Slurm Environment Variable | Description of the Variable |
|---|---|
| SLURM_NTASKS | Number of tasks specified with -n |
| SLURM_CPUS_PER_TASK | Number of CPU cores specified with -c |
| SLURM_JOB_ID | Job ID |
| SLURM_MEM_PER_CPU | Memory allocated with --mem-per-cpu |
How to know which compute nodes have been allocated for parallel job
To confirm that a job has been queued, use the squeue command. The displayed items include NODES, indicating the number of allocated nodes, and NODELIST, showing the list of node names.
xxxxx-pg@at022vm02:~$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
642 parabrick test.sh xxxxx-pg R 0:23 3 igt[010,015-016]
Alternatively, you can use the pestat command to check the deployment status of jobs to compute nodes.
yxxxx-pg@at022vm02:~/parabricks$ pestat
Hostname Partition Node Num_CPU CPUload Memsize Freemem Joblist
State Use/Tot (15min) (MB) (MB) JobID User ...
igt009 igt009 down* 0 48 0.03 386452 366715
igt010 parabricks mix 4 48 0.24* 386462 377973 780 yxxxx-pg
igt015 igt015 idle 0 48 0.00 386458 380937
igt016 parabricks* idle 0 48 0.17 386462 379696
This guide detailed how to handle CPU and memory allocations for parallel jobs in Slurm, including how to execute specific job types and check which compute nodes have been allocated. It covered executing MPI and OpenMP programs, as well as the use of Slurm environment variables to direct the parallel operations internally.
Notes on Running Parallel Programs in the Personal Genome Analysis Section
When using compute nodes equipped with GPUs in the personal genome analysis section, even if a job occupies all installed GPUs, other jobs that do not use GPUs might still be submitted to the same compute node if CPU and memory resources are available. To prevent this and ensure exclusive operation of parallel jobs, the following execution method is recommended.
Executing MPI Parallel Programs
#!/bin/bash
#SBATCH -N 1-1
#SBATCH --exclusive
#SBATCH -n 8
#SBATCH --gres=gpu:4
#SBATCH -o %x.%J.out
#SBATCH --job-name my_program
mpirun mpi_test
The above settings request the following from Slurm:
-N 1-1requests the allocation of one node.--exclusiverequests exclusive allocation of the assigned node.-n 8requests 8 parallel operations within the node. mpirun recognizes this number as the MPI parallelism count. (Adjust the parallel count as necessary.)
Even with --exclusive specified, please ensure to specify -n. Without it, the parallel count may not be recognized correctly. The --exclusive option allocates 48 CPU cores and 192GB of memory. The parallel count for the MPI program is recognized as 8, as specified by -n.
MPI Sample Program (for verifying parallel count, not utilizing GPUs)
#include <mpi.h>
#include <stdio.h>
int main(int argc, char** argv) {
MPI_Init(NULL, NULL);
int world_size;
MPI_Comm_size(MPI_COMM_WORLD, &world_size);
int world_rank;
MPI_Comm_rank(MPI_COMM_WORLD, &world_rank);
char processor_name[MPI_MAX_PROCESSOR_NAME];
int name_len;
MPI_Get_processor_name(processor_name, &name_len);
printf("Hello world from processor %s, rank %d out of %d processors\n",
processor_name, world_rank, world_size);
MPI_Finalize();
}
The allocation of CPU cores when executing the above program with the submission script is illustrated below.
yxxxx-pg@at022vm02:~/mpitest$ pestat
Hostname Partition Node Num_CPU CPUload Memsize Freemem Joblist
State Use/Tot (15min) (MB) (MB) JobID User ...
igt009 igt009 down* 0 48 0.03 386452 366715
igt010 parabricks alloc 48 48 0.25* 386462 377564 1663 yxxxx-pg
igt015 igt015 idle 0 48 0.00 386458 380819
igt016 igt016 idle 0 48 0.05 386462 379490
This shows that on compute node igt010, the job operates with all 48 available CPU cores allocated, not just the parallel count of 8 specified by -n. The output of the sample program demonstrates it operates in 8 parallel.
Executing OpenMP Thread Parallel Programs
To execute an OpenMP thread parallel program while occupying a compute node, describe the directive lines as follows.
Job Script Specifications
#!/bin/bash
#SBATCH -N 1-1
#SBATCH -n 1
#SBATCH --gres=gpu:4
#SBATCH --exclusive
omp_sample
-N 1-1specifies one compute node.-n 1specifies one task.--exclusivespecifies the exclusive use of one compute node.
Specifying --exclusive results in the physical compute node being recognized as having all available cores (48 cores), with the node being occupied. The allocated memory, by configuration, is 192GB.
Thread Parallel Sample Program (for verifying parallel count, not utilizing GPUs)
#include <stdio.h>
#include <omp.h>
int main()
{
printf("hello openmp world \n");
#pragma omp parallel
{
printf("processing thread num= %d \n", omp_get_thread_num());
}
printf("good by \n");
return 0;
}
The marked lines operate in thread parallel.
Output of the Sample Program (abbreviated for brevity)
hello openmp world
processing thread num= 27
...
processing thread num= 0
...
good by
It can be seen operating in 48 thread parallel.
If you wish to occupy a node and explicitly specify the thread parallel count, specify -c and set the environment variable O
OMP_NUM_THREADS environment variable.
Job Script Specifications
#!/bin/bash
#SBATCH -N 1-1
#SBATCH -n 1
#SBATCH --exclusive
#SBATCH -c 8
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
omp_sample
Specifying --exclusive results in the job operating with all CPU cores of the compute node being allocated. The marked lines are crucial for ensuring that the compute node's resources are dedicated to the job, and the program operates within the specified core count.